Detailed results of SGR42_XRay_em_bcr3 by PSVS
Output from PDBStat
Output from PROCHECK
Ramachandran Plot for all models
Text summary of Ramachandran Plot
+----------<<< P R O C H E C K S U M M A R Y >>>----------+
| |
| SGR42_XRay_em_bcr3_noHs_000.rin 0.0 112 residues |
| |
| Ramachandran plot: 90.8% core 9.2% allow 0.0% gener 0.0% disall |
| |
| All Ramachandrans: 0 labelled residues (out of 108) |
+| Chi1-chi2 plots: 1 labelled residues (out of 64) |
JPEG image for all model Ramachandran Plot

Residue Properties for all models
JPEG for all model Residue Properties - page $num_n

JPEG for all model Residue Properties - page $num_n

Model Secondary Structures from Procheck
JPEG for Model Secondary Structures - page $num_n

JPEG for Model Secondary Structures - page $num_n

Ramachandran Plots for each residue
JPEG for residue Ramachandran Plots - page $num_n

JPEG for residue Ramachandran Plots - page $num_n

JPEG for residue Ramachandran Plots - page $num_n

JPEG for residue Ramachandran Plots - page $num_n

JPEG for residue Ramachandran Plots - page $num_n

JPEG for residue Ramachandran Plots - page $num_n

Ramachandran analysis for each residue from Molprobity
Chi1-Chi2 Plots for each residue
JPEG for residue Chi1-Chi2 Plots - page $num_n

JPEG for residue Chi1-Chi2 Plots - page $num_n

JPEG for residue Chi1-Chi2 Plots - page $num_n

JPEG for residue Chi1-Chi2 Plots - page $num_n

Procheck G-factors for phi-psi for each residue
JPEG image for residue phi-psi G-factors
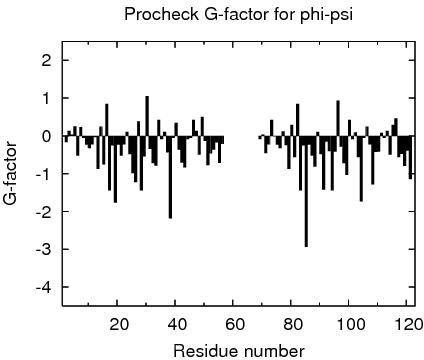
Table of Procheck G-factors for phi-psi for ordered residues
#phipsi_gfactor
#Residue\Model average
2 -0.16
3 0.13
4 0.03
5 0.25
6 -0.52
7 0.23
8 -0.05
9 -0.23
10 -0.32
11 -0.22
12 -0.03
13 -0.87
14 0.24
15 -0.75
16 0.84
17 -1.44
18 -0.25
19 -1.76
20 -0.23
21 -0.52
22 -0.22
23 0.10
24 -0.48
25 -0.98
26 -1.22
27 0.38
28 -1.44
29 -0.54
30 1.05
31 -0.34
32 -0.72
33 -0.78
34 0.42
35 -0.08
36 0.10
37 -0.43
38 -2.18
39 -0.05
40 0.34
41 -0.36
42 -0.70
43 -0.83
44 -0.08
45 -0.05
46 0.42
47 0.13
48 -0.49
49 0.50
50 -0.13
51 -0.77
52 -0.46
53 -0.36
54 -0.17
55 -0.71
56 -0.21
69 -0.08
70 0.03
71 -0.45
72 -0.22
73 0.42
74 -0.01
75 -0.23
76 -0.32
77 0.12
78 -0.24
79 -0.87
80 0.29
81 -0.56
82 0.84
83 -1.44
84 -0.25
85 -2.93
86 -0.23
87 -0.52
88 -0.81
89 0.10
90 -0.48
91 -1.42
92 -0.15
93 -0.40
94 -1.44
95 -0.41
96 0.93
97 -0.28
98 -0.72
99 -1.03
100 0.42
101 -0.08
102 0.09
103 -0.56
104 -1.73
105 -0.05
106 0.24
107 -0.22
108 -1.28
109 -0.42
110 -0.41
111 0.08
112 -0.05
113 0.13
114 -0.49
115 0.29
116 0.46
117 -0.56
118 -0.47
119 -0.79
120 -0.39
121 -1.14
#Reported_Model_Average -0.339
#Overall_Average_Reported -0.339
Procheck G-factors for all dihedral angles for each residue
JPEG image for residue all dihedral G-factors
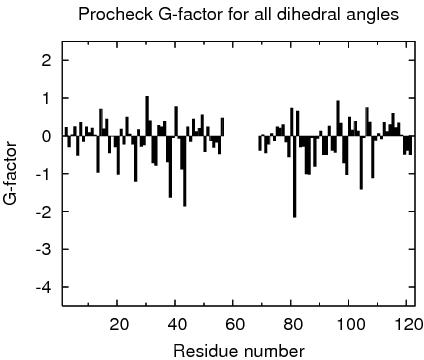
Table of Procheck G-factors for all dihedrals for ordered residues
#alldih_gfactor
#Residue\Model average
1 0.00
2 0.23
3 -0.29
4 0.03
5 0.25
6 -0.52
7 0.36
8 -0.15
9 0.24
10 0.09
11 0.21
12 0.02
13 -0.97
14 0.71
15 0.19
16 0.45
17 -0.45
18 -0.01
19 -0.29
20 -1.02
21 0.18
22 -0.22
23 0.50
24 0.05
25 -0.22
26 -1.21
27 0.17
28 -0.28
29 -0.24
30 1.05
31 0.40
32 -0.72
33 -0.78
34 0.28
35 0.24
36 0.39
37 -0.69
38 -1.63
39 -0.05
40 0.78
41 -0.07
42 -0.88
43 -1.86
44 0.24
45 -0.15
46 0.45
47 0.12
48 0.20
49 0.56
50 -0.42
51 0.24
52 -0.13
53 -0.31
54 -0.17
55 -0.48
56 0.47
57 0.00
68 0.61
69 -0.39
70 0.03
71 -0.45
72 -0.22
73 0.06
74 -0.13
75 0.24
76 0.21
77 0.30
78 -0.16
79 -0.56
80 0.74
81 -2.15
82 0.66
83 -0.29
84 -0.28
85 -1.01
86 -1.02
87 -0.05
88 -0.81
89 -0.07
90 0.13
91 -0.50
92 -0.50
93 0.26
94 -0.39
95 -0.44
96 0.93
97 0.34
98 -0.72
99 -1.03
100 0.50
101 0.16
102 0.39
103 0.13
104 -1.41
105 -0.05
106 0.75
107 0.37
108 -1.11
109 -0.12
110 0.06
111 -0.08
112 0.36
113 0.12
114 0.30
115 0.60
116 0.22
117 0.35
118 0.02
119 -0.49
120 -0.39
121 -0.50
122 -2.16
#Reported_Model_Average -0.123
#Overall_Average_Reported -0.123
Output from Verify3D
Verify3D Score over a window of $winsize_s residues
JPEG image for Verify3D Score
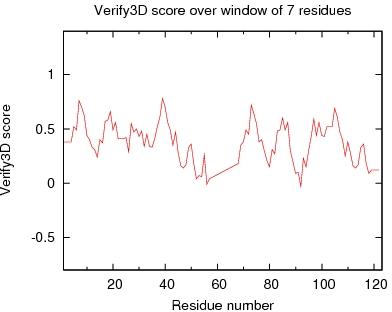
Table of Verify3D scores for ordered residues across all models
#verify3d
#Residue\Model only_model
1 0.00
2 0.93
3 -0.84
4 0.44
5 1.10
6 0.49
7 0.55
8 1.00
9 0.71
10 1.00
11 0.08
12 0.51
13 -0.74
14 0.23
15 0.51
16 0.55
17 0.52
18 1.25
19 0.24
20 0.71
21 0.28
22 1.10
23 -0.68
24 1.00
25 0.25
26 0.24
27 0.66
28 0.34
29 0.23
30 1.10
31 0.47
32 0.49
33 -0.25
34 1.00
35 -0.68
36 1.04
37 0.28
38 0.41
39 1.10
40 0.51
41 1.62
42 0.51
43 0.47
44 -0.68
45 -0.09
46 0.08
47 1.40
48 0.24
49 -0.33
50 0.34
51 -0.46
52 1.06
53 0.28
54 0.14
55 -0.74
56 -0.10
57 0.25
68 0.93
69 -0.84
70 0.64
71 1.10
72 0.49
73 0.08
74 1.00
75 0.71
76 1.00
77 0.08
78 0.51
79 -0.74
80 0.23
81 0.34
82 0.08
83 0.52
84 1.25
85 0.24
86 0.71
87 0.28
88 1.10
89 -0.68
90 1.00
91 -0.57
92 -0.41
93 -0.09
94 0.34
95 0.23
96 1.10
97 0.47
98 0.49
99 0.49
100 1.00
101 -0.68
102 1.04
103 0.28
104 0.41
105 1.10
106 0.51
107 0.96
108 0.51
109 0.47
110 -0.68
111 -0.09
112 0.08
113 1.40
114 0.24
115 -0.33
116 0.34
117 -0.46
118 1.06
119 0.28
120 0.14
121 -0.40
122 -0.10
#Reported_Model_Average 0.369
#Overall_Average_Reported 0.369
Output from ProsaII
ProsaII Score over a window of $winsize_s residues
JPEG image for ProsaII Score
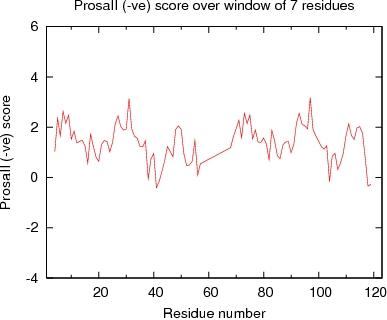
Table of Verify3D scores for ordered residues across all models
#verify3d
#Residue\Model only_model
1 0.00
2 0.93
3 -0.84
4 0.44
5 1.10
6 0.49
7 0.55
8 1.00
9 0.71
10 1.00
11 0.08
12 0.51
13 -0.74
14 0.23
15 0.51
16 0.55
17 0.52
18 1.25
19 0.24
20 0.71
21 0.28
22 1.10
23 -0.68
24 1.00
25 0.25
26 0.24
27 0.66
28 0.34
29 0.23
30 1.10
31 0.47
32 0.49
33 -0.25
34 1.00
35 -0.68
36 1.04
37 0.28
38 0.41
39 1.10
40 0.51
41 1.62
42 0.51
43 0.47
44 -0.68
45 -0.09
46 0.08
47 1.40
48 0.24
49 -0.33
50 0.34
51 -0.46
52 1.06
53 0.28
54 0.14
55 -0.74
56 -0.10
57 0.25
68 0.93
69 -0.84
70 0.64
71 1.10
72 0.49
73 0.08
74 1.00
75 0.71
76 1.00
77 0.08
78 0.51
79 -0.74
80 0.23
81 0.34
82 0.08
83 0.52
84 1.25
85 0.24
86 0.71
87 0.28
88 1.10
89 -0.68
90 1.00
91 -0.57
92 -0.41
93 -0.09
94 0.34
95 0.23
96 1.10
97 0.47
98 0.49
99 0.49
100 1.00
101 -0.68
102 1.04
103 0.28
104 0.41
105 1.10
106 0.51
107 0.96
108 0.51
109 0.47
110 -0.68
111 -0.09
112 0.08
113 1.40
114 0.24
115 -0.33
116 0.34
117 -0.46
118 1.06
119 0.28
120 0.14
121 -0.40
122 -0.10
#Reported_Model_Average 0.369
#Overall_Average_Reported 0.369
Output from MolProbity
VdW violations from MAGE
JPEG image for MAGE VdW violation
Table of MAGE VdW violations for ordered residues across all models
#mage_clash
#Residue\Model only_model
1.000 0
2.000 1
3.000 0
4.000 0
5.000 0
6.000 0
7.000 0
8.000 0
9.000 1
10.000 0
11.000 0
12.000 1
13.000 0
14.000 0
15.000 1
16.000 0
17.000 1
18.000 0
19.000 0
20.000 1
21.000 0
22.000 0
23.000 0
24.000 1
25.000 0
26.000 0
27.000 0
28.000 0
29.000 0
30.000 0
31.000 0
32.000 0
33.000 0
34.000 0
35.000 0
36.000 0
37.000 0
38.000 2
39.000 0
40.000 0
41.000 0
42.000 0
43.000 0
44.000 0
45.000 0
46.000 0
47.000 0
48.000 0
49.000 0
50.000 0
51.000 0
52.000 0
53.000 0
54.000 0
55.000 1
56.000 0
57.000 0
58.000 0
59.000 0
60.000 0
61.000 0
62.000 0
63.000 0
64.000 0
65.000 0
66.000 0
67.000 0
68.000 0
69.000 0
70.000 0
71.000 0
72.000 0
73.000 0
74.000 1
75.000 0
76.000 0
77.000 0
78.000 0
79.000 0
80.000 0
81.000 0
82.000 0
83.000 0
84.000 1
85.000 1
86.000 0
87.000 1
88.000 0
89.000 0
90.000 0
91.000 0
92.000 0
93.000 0
94.000 0
95.000 0
96.000 0
97.000 0
98.000 0
99.000 0
100.000 0
101.000 0
102.000 0
103.000 0
104.000 2
105.000 0
106.000 0
107.000 0
108.000 0
109.000 0
110.000 0
111.000 0
112.000 0
113.000 0
114.000 0
115.000 0
116.000 0
117.000 0
118.000 0
119.000 0
120.000 0
121.000 0
122.000 6
#Reported_Model_Average 0.180
#Overall_Average_Reported 0.180
List of bad contacts calculated by MAGE
/farm/software/bin/probe
: 1776:M 122 LYS H :M 122 LYS 1HD : -0.944: 48
: 1776:M 122 LYS 1HD :M 122 LYS N : -0.758: 48
: 1776:M 122 LYS H :M 122 LYS CD : -0.693: 48
: 1776:M 104 ASN C :M 104 ASN 2HD2 : -0.523: 19
: 1776:M 12 ASN 2HB :M 15 ASP OD2 : -0.512: 28
: 1776:M 85 ARG 1HB :M 84 TYR O : -0.505: 14
: 1776:M 38 ASN 2HD2 :M 38 ASN C : -0.474: 21
: 1776:M 17 TYR O :M 20 PHE 1HB : -0.428: 11
: 1776:M 24 VAL 1HG2 :M 2 ILE 3HG2 : -0.421: 29
: 1776:M 9 ARG NH2 :M 55 VAL 1HG1 : -0.412: 31
: 1776:M 87 GLU HA :M 74 VAL O : -0.411: 13
#sum2 ::6.19 clashscore : 4.69 clashscore B<40
#summary::1776 atoms:1705 atoms B<40:198417 potential dots:12400.0 A^2:11 bumps:8 bumps B<40:599 score
Output from PDB validation software
Summary from PDB validation
May. 10, 21:37:52 2013
Greetings,
[ Text modified to reflect that this was run under PSVS - Aneerban Bhattacharya: Dec 2005 ]
The following checks were made on :
-----------------------------------------
DISTANCES AND ANGLES
We have checked your intra and intermolecular distances and angles with the
procedures currently in place at PDB:
==> The following solvent molecules are further away than 3.5 Angstroms from
macromolecule atoms which are available for hydrogen bonding in the
asymmetric unit.
none
The coordinates for water molecules which could be translated back into
the asymmetric unit are listed. If you do not indicate otherwise we will
replace the solvent coordinates in the entry with the ones below:
none
==> Close contacts in same asymmetric unit. Distances smaller than 2.2
Angstroms are considered as close contacts.
none
==> Close contacts based on crystal symmetry. Distances smaller than 2.2
Angstroms are considered as close contacts.
none
==> Bond and angle checks are performed by first computing the average rms
error for all bonds and angles relative to standard values for nucleotide
units [L. Clowney et al., Geometric Parameters in Nucleic Acids: Nitrogenous
Bases, J.Am.Chem.Soc. 1996, 118, 509-518; A. Gelbin et al., Geometric
Parameters in Nucleic Acids: Sugar and Phosphate Constituents, J.Am.Chem.Soc.
1996, 118, 519-529] and amino acid units [R.A. Engh and R. Huber, Accurate
Bond and Angle Parameters for X-ray protein structure refinement, Acta
Crystallogr. 1991, A47, 392-400]. Any bond or angle which deviates from the
dictionary values by more than six times this computed rms error is
identified as an outlier.
*** Covalent Bond Lengths:
The RMS deviation for covalent bonds relative to the standard
dictionary is 0.004 Angstroms
All covalent bonds lie within a 6.0*RMSD range about the
standard dictionary values.
*** Covalent Angle Values:
The RMS deviation for covalent angles relative to the standard
dictionary is 1.2 degrees.
The following table contains a list of the covalent bond angles
greater than 6.0*RMSD.
Deviation Residue Chain Sequence AT1 - AT2 - AT3 Bond Dictionary
Name ID Number Angle Value
--------------------------------------------------------------------------------
-10.9 ARG A 48 N - CA - C 100.3 111.2
-8.7 ARG B 48 N - CA - C 102.5 111.2
TORSION ANGLES
The torsion angle distributions have been checked. The postscript file of the
conformation rings showing the torsion angle distributions will be sent in a
separate E-mail message.
CHIRALITY
The chirality has been checked and there are no incorrect carbon chiral centers.
Some of O1P and O2P atoms do not follow the convention defined in the standard
IUBMB nomenclature (Liebecq, C. Compendium of Biochemical Nomenclature and Related
Documents, 2nd ed.; Portland Press: London and Chapel Hill, 1992). If you do not
indicate otherwise, we will switch the labels of O1P and O2P as shown below.
OTHER IMPORTANT ISSUES
==> Please check carefully REMARKS 3 and 200 and fill in the parameters as
appropriate.
==> The following residues have missing atoms:
RES MOD#C SEQ ATOMS
MET( A 1) CG SD CE
SGR42_XRay_em_bcr3.pdb: Error: Z value is 6. It should be 3.