| NESG ID: | NAME |
| PDB ID: | |
| Deposition date: | |
| Common Name: | |
| Class: | |
| Length (a.a.): | 50 |
| Organism: | |
| SwissProt / TrEMBL ID: | |
| # models: | 20 |
| Oligomerization: | monomer |
| Molecular weight: | 5669 |
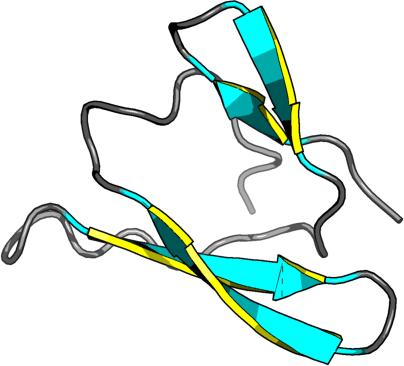  | |
 |
Analyses performed for user defined residues.
The constraints analysis is based on the following files: NOE distance constraints file. Angular constraints file. H-bond constraints file.
Procheck analysis,RMSD calculation and structure superimposition are based on: User defined residues
|
|
Secondary Structure Elements:
alpha helices:
beta strands: 11T-14T, 2R-6R, 44L-46L, 19U-21U, 26U-30U, 36A-40A
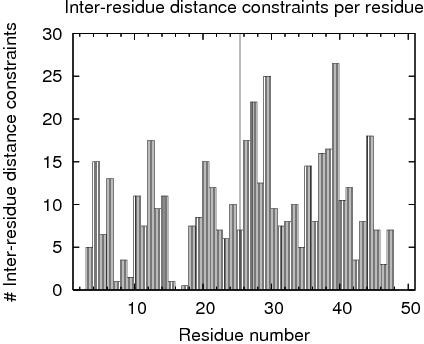
| Total number of restricting constraints per restrained residue: | 11.0 |
| Restricting long range constraints per restrained residue: | 5.0 |
Distance violations per model
Calculated using sum over r^-6
| 0.1 - 0.2 Å | 0.2 - 0.5 Å | > 0.5 Å |
| 5.6 | 13.95 | 22.35 |
Dihedral angle violations per model
1 - 10 ° > 10 ° 1.2 4.5
FIDs deposited in the BMRB? no
| RMSD | All residues | Ordered residues2 | Selected residues3 |
| All backbone atoms | 1.2 Å | 0.7 Å | 0.7 Å |
| All heavy atoms | 2.0 Å | 1.1 Å | 1.2 Å |
Ramachandran Plot Summary for selected residues3 from Procheck
| Most favoured regions | Additionally allowed regions | Generously allowed regions | Disallowed regions |
| 88.9% | 10.8% | 0.1% | 0.1% |
Ramachandran Plot Summary for selected residues3 from Richardson Lab's Molprobity
| Most favoured regions | Allowed regions | Disallowed regions | View plot View model summary |
| 98.8% | 1% | 0.2% |
Global quality scores
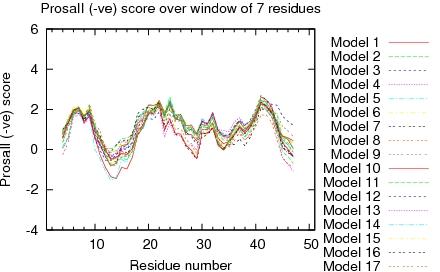
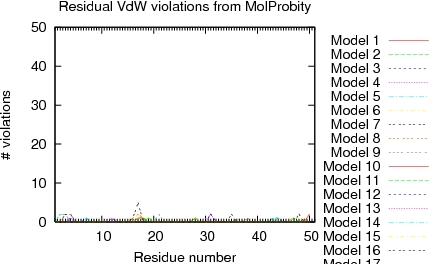
| Program | Verify3D | ProsaII (-ve) | Procheck (phi-psi)3 | Procheck (all)3 | MolProbity Clashscore |
| -Raw score | 0.26 | 0.44 | -0.37 | 0.06 | 2.54 |
| Z-score1 | -3.21 | -0.87 | -1.14 | 0.35 | 1.09 |
Close Contacts and Deviations from Ideal Geometry (from PDB validation software)
| Number of close contacts (within 1.6 Å for H atoms, 2.2 Å for heavy atoms): | 0 |
| RMS deviation for bond angles: | 0.8 ° |
| RMS deviation for bond lengths: | 0.011 Å |
1 With respect to mean and standard deviation for a set of 252 X-ray structures < 500 residues, of resolution <= 1.80 Å, R-factor <= 0.25 and R-free <= 0.28; a positive value indicates a 'better' score
2Order residues: 4A-14A,18A-46A
3Selected residues: 4A-14A,17A-46A
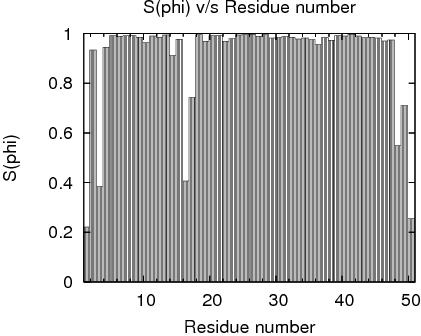
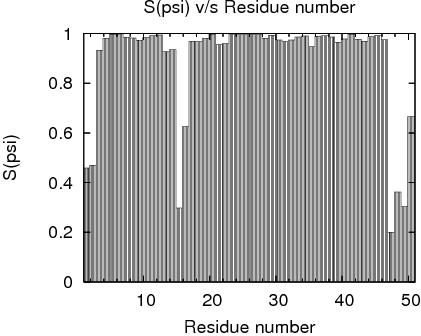
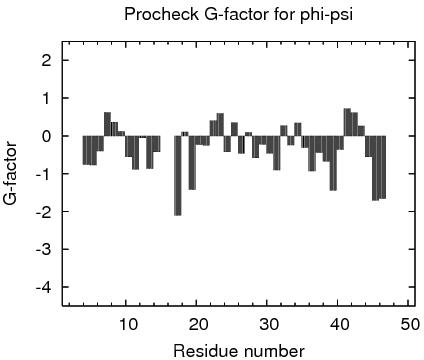
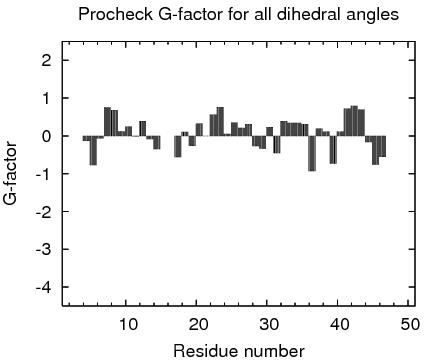
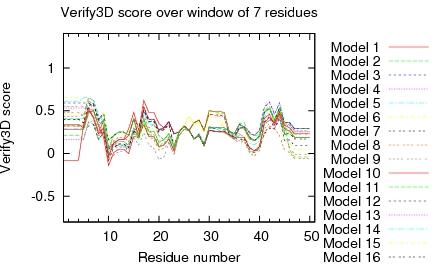
Residue Plot of Ramachandran anlysis(based on data from Richardson Lab's Molprobity)
References:
1. Luthy R, Bowie J U and Eisenberg D, "Assessment of protein models with three-dimensional profiles", Nature 356 (1992): 83-85
2. Bowie J U, Luthy R and Eisenberg D, "A Method to Identify Protein Sequences that Fold into a Known Three-Dimensional Structure", Science 253 (1991): 164-169
3. Sippl M J, "Recognition of Errors in Three-Dimensional Structures of Proteins", Proteins 17 (1993): 355-362
4. Sippl M J, "Calculation of Conformation Ensembles from Potentials of Mean Force", J Mol Biol 213 (1990): 859-883
5. Laskowski R Ai et al, "AQUA and PROCHECK_NMR: Programs for checking the quality of proteins structures solved by NMR", J Biomolec NMR 8 (1996): 477-486
6. Laskowski R A et al "PROCHECK: a program to check the stereochemical quality of protein structures" J Appl Cryst, 26 (1993): 283-291
7. Word J M et al, "Exploring steric constrains on protein mutations using MAGE / PROBE", Prot Sci 9 (2000): 2251-2259
8. Word J M et al, "Asparagine and Glutamine: Using Hydrogen Atom Contacts in the Choice of Side-chain Amide Orientation", J Mol Biol 285 (1999): 1735-1747
9. Word J M et al, "Visualizing and Quantifying Molecular Goodness-of-Fit: Small-probe Contact Dots with Explicit Hydrogens", J Mol Biol 285 (1999): 1711-1733
10. Tejero R and Montelione G T, "PDBStat", unpublished
11. Luthy R, McLachlan A D and Eisenberg D, "Secondary Structure-Based Profiles: Use of Structure-Conserving Scoring Tables in Searching Protein Sequence Databases for Structural Similarities", Proteins 10 (1991): 229-239
12. Richardson D C, Richardson J S, "The kinemage: a tool for scientific communication", Prot Sci 1(1) (1992): 3-9
13. Koradi, R, et al, "MOLMOL: a program for display and analysis of macromolecular structures ", J Mol Graphics 14 (1996): 51-55.
14. Güntert, P, Mumenthaler, C & Wüthrich, K "Torsion angle dynamics for NMR structure calculation with the new program DYANA", J. Mol. Biol 273 (1997): 283-298
15. Lovell S C et al, "Structure validation by Calpha geometry: phi,psi and Cbeta deviation" Proteins (2003) 50: 437-450
16. Kabsch W, Sander C, "Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features", Biopolymers (1983) 22: 2577-2637
17. Bagaria, A., Jaravine, V., Huang, Y.J., Montelione, G.T., and Guntert, P. "Protein structure validation by generalized linear model root-mean-square deviation prediction". Protein Sci 21(2012), 229-238.